MAGIC - Markov Affinity-based Graph Imputation of Cells¶






Markov Affinity-based Graph Imputation of Cells (MAGIC) is an algorithm for denoising high-dimensional data most commonly applied to single-cell RNA sequencing data. MAGIC learns the manifold data, using the resultant graph to smooth the features and restore the structure of the data.
To see how MAGIC can be applied to single-cell RNA-seq, elucidating the epithelial-to-mesenchymal transition, read our publication in Cell.
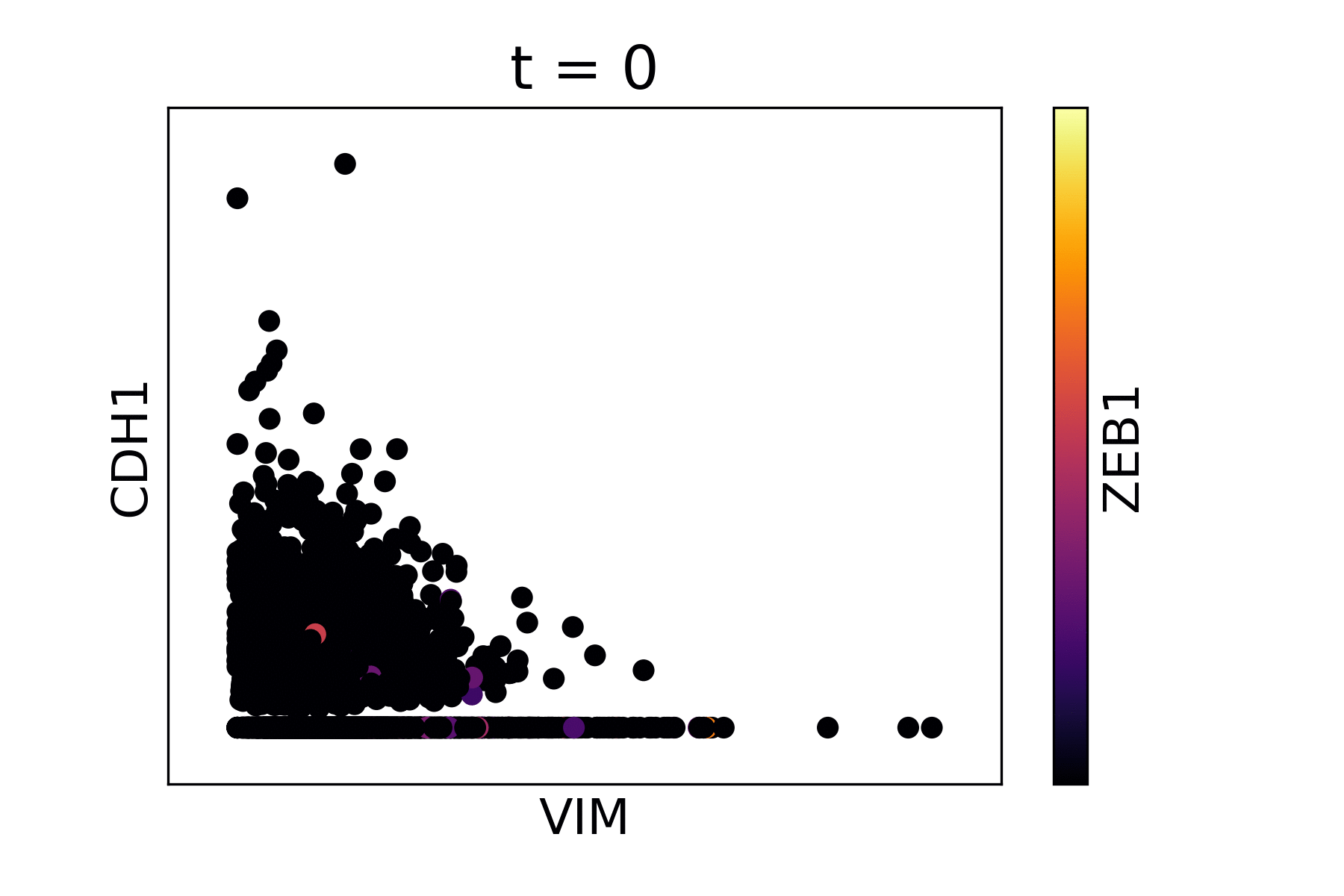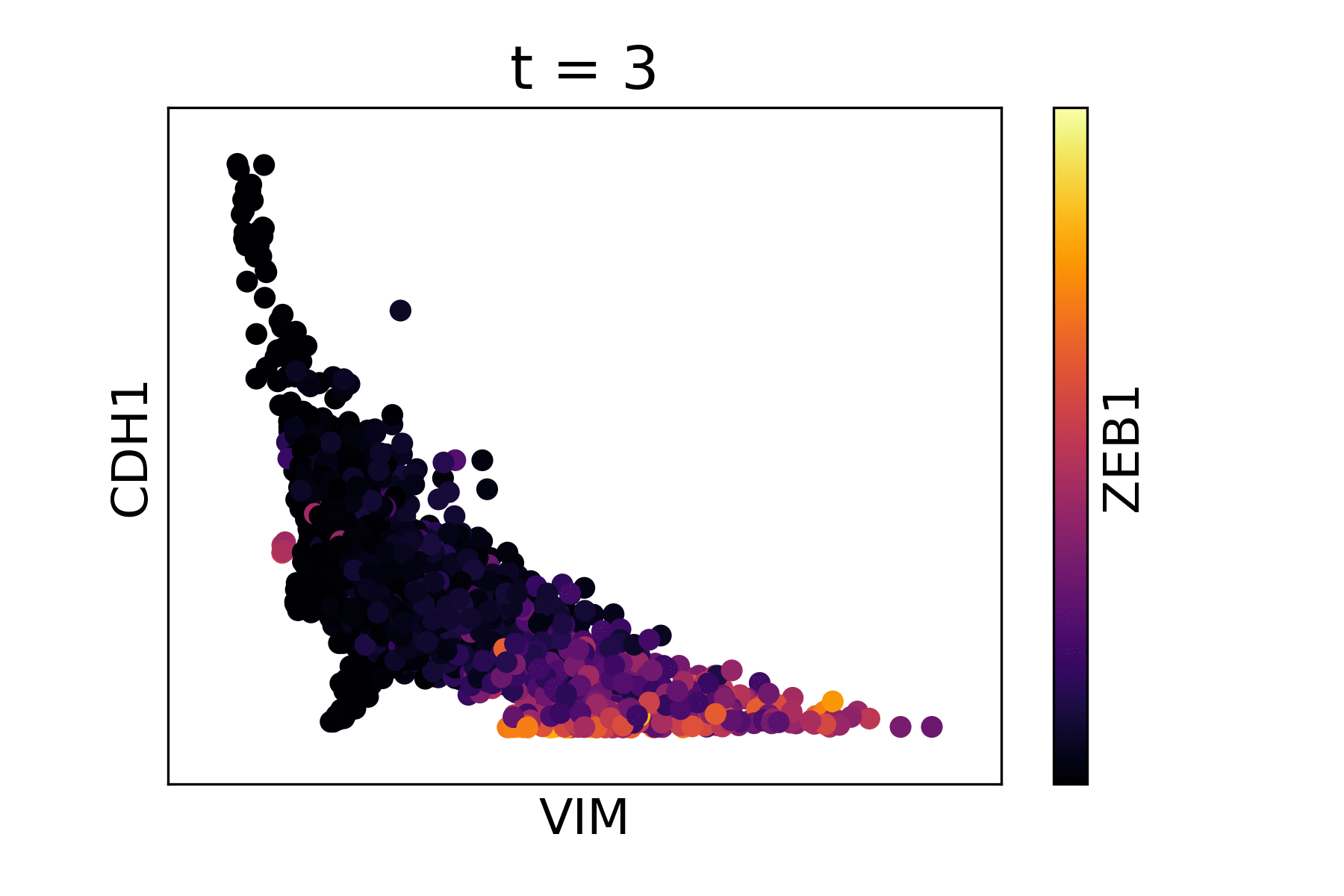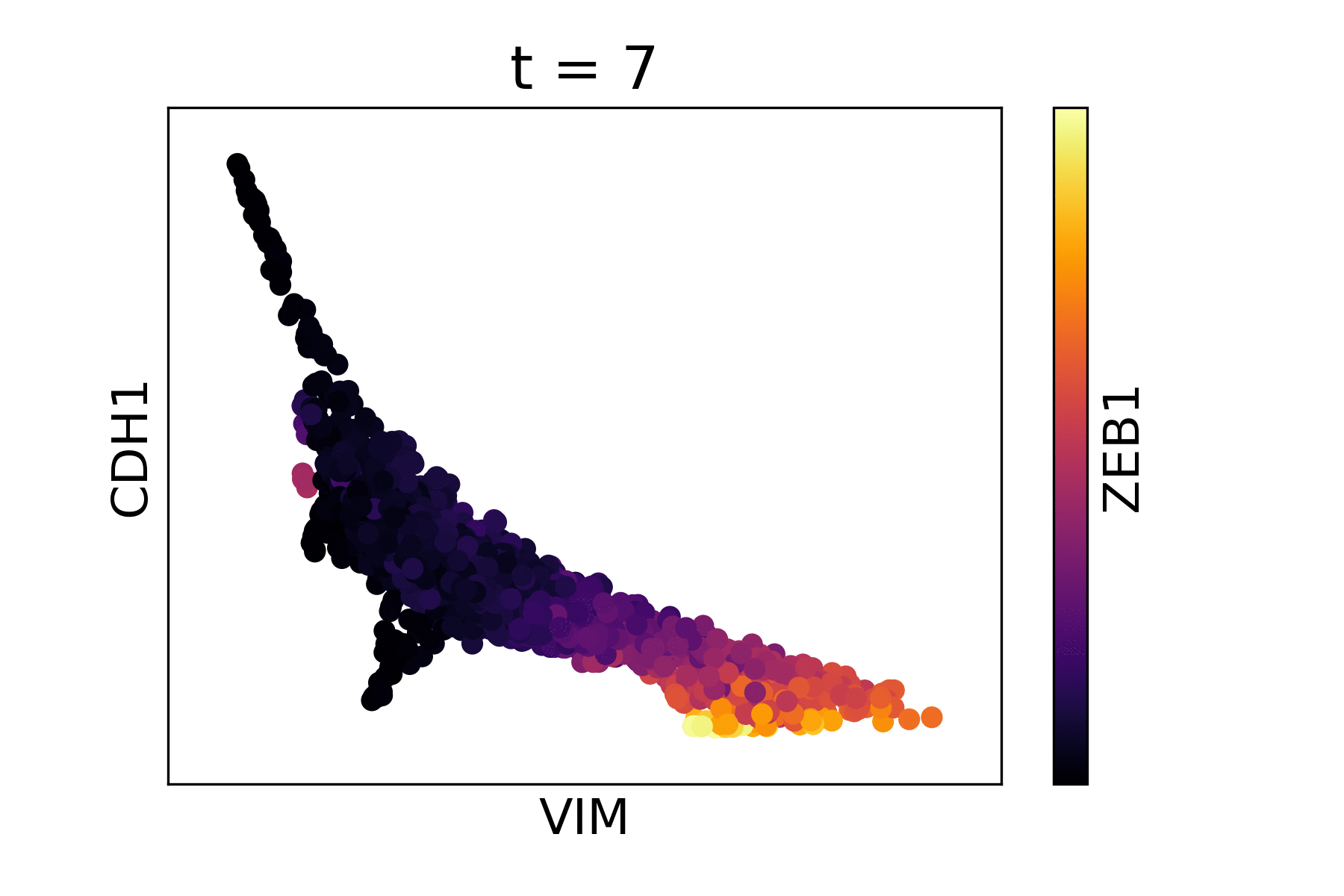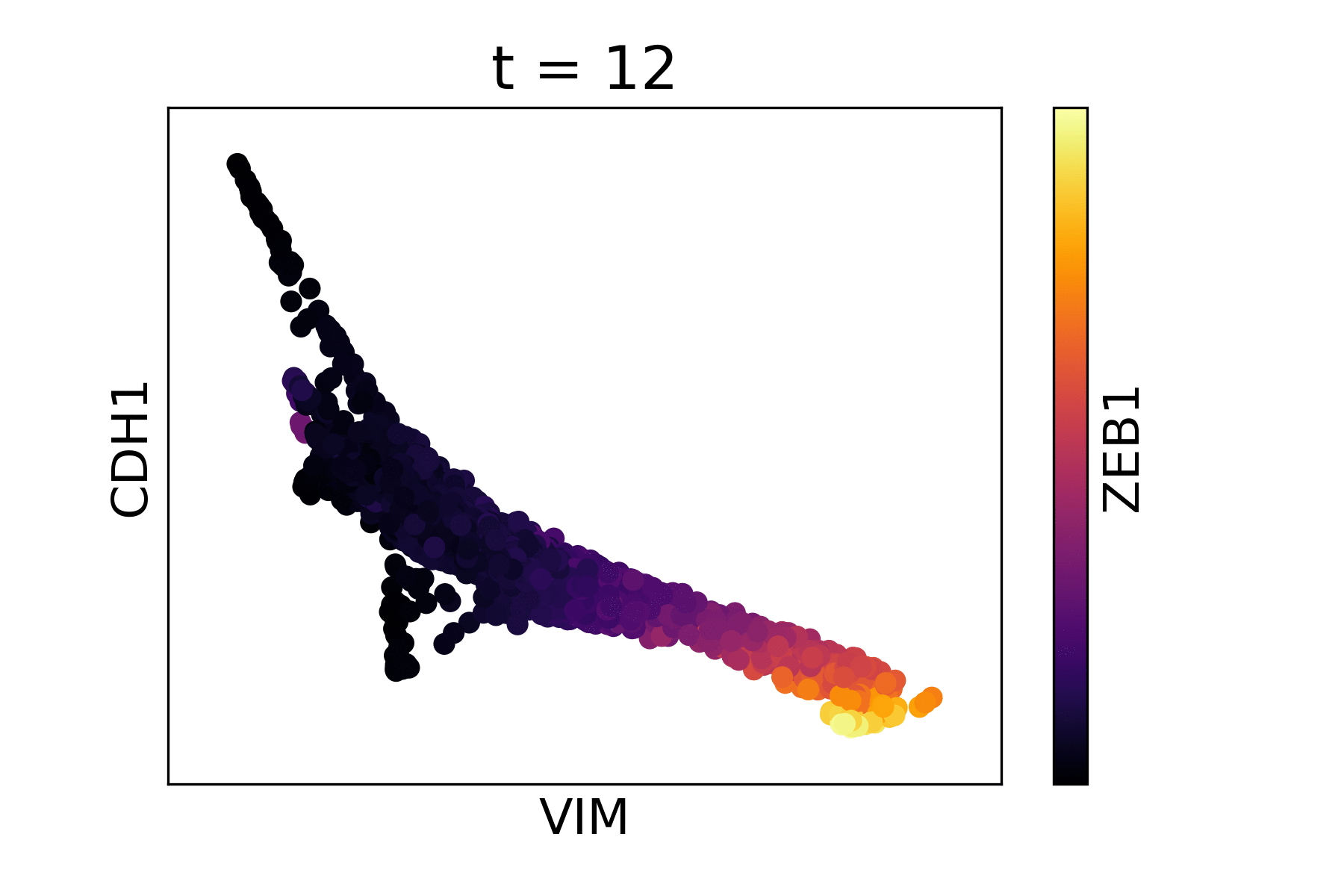
Magic reveals the interaction between Vimentin (VIM), Cadherin-1 (CDH1), and Zinc finger E-box-binding homeobox 1 (ZEB1, encoded by colors).
Quick Start¶
To run MAGIC on your dataset, create a MAGIC operator and run fit_transform. Here we show an example with a small, artificial dataset located in the MAGIC repository:
import magic
import pandas as pd
import matplotlib.pyplot as plt
X = pd.read_csv("MAGIC/data/test_data.csv")
magic_operator = magic.MAGIC()
X_magic = magic_operator.fit_transform(X, genes=['VIM', 'CDH1', 'ZEB1'])
plt.scatter(X_magic['VIM'], X_magic['CDH1'], c=X_magic['ZEB1'], s=1, cmap='inferno')
plt.show()
magic.plot.animate_magic(X, gene_x='VIM', gene_y='CDH1', gene_color='ZEB1', operator=magic_operator)
Help¶
If you have any questions or require assistance using MAGIC, please contact us at https://krishnaswamylab.org/get-help
-
class
magic.MAGIC(knn=5, knn_max=None, decay=1, t=3, n_pca=100, solver='exact', knn_dist='euclidean', n_jobs=1, random_state=None, verbose=1)[source] MAGIC operator which performs dimensionality reduction.
Markov Affinity-based Graph Imputation of Cells (MAGIC) is an algorithm for denoising and transcript recover of single cells applied to single-cell RNA sequencing data, as described in van Dijk et al, 2018 [1].
Parameters: - knn (int, optional, default: 5) – number of nearest neighbors from which to compute kernel bandwidth
- knn_max (int, optional, default: None) – maximum number of nearest neighbors with nonzero connection. If None, will be set to 3 * knn
- decay (int, optional, default: 1) – sets decay rate of kernel tails. If None, alpha decaying kernel is not used
- t (int, optional, default: 3) – power to which the diffusion operator is powered. This sets the level of diffusion. If ‘auto’, t is selected according to the Procrustes disparity of the diffused data
- n_pca (int, optional, default: 100) – Number of principal components to use for calculating neighborhoods. For extremely large datasets, using n_pca < 20 allows neighborhoods to be calculated in roughly log(n_samples) time.
- solver (str, optional, default: 'exact') – Which solver to use. “exact” uses the implementation described in van Dijk et al. (2018) [1]. “approximate” uses a faster implementation that performs imputation in the PCA space and then projects back to the gene space. Note, the “approximate” solver may return negative values.
- knn_dist (string, optional, default: 'euclidean') – Distance metric for building kNN graph. Recommended values: ‘euclidean’, ‘cosine’. Any metric from scipy.spatial.distance can be used. Custom distance functions of form f(x, y) = d are also accepted
- n_jobs (integer, optional, default: 1) – The number of jobs to use for the computation. If -1 all CPUs are used. If 1 is given, no parallel computing code is used at all, which is useful for debugging. For n_jobs below -1, (n_cpus + 1 + n_jobs) are used. Thus for n_jobs = -2, all CPUs but one are used
- random_state (integer or numpy.RandomState, optional, default: None) – The generator used to initialize random PCA If an integer is given, it fixes the seed Defaults to the global numpy random number generator
- verbose (int or boolean, optional (default: 1)) – If True or > 0, print status messages
-
X¶ Input data
Type: array-like, shape=[n_samples, n_features]
-
X_magic¶ Output data
Type: array-like, shape=[n_samples, n_features]
-
graph¶ The graph built on the input data
Type: graphtools.BaseGraph
Examples
>>> import magic >>> import pandas as pd >>> import matplotlib.pyplot as plt >>> X = pd.read_csv("../../data/test_data.csv") >>> X.shape (500, 197) >>> magic_operator = magic.MAGIC() >>> X_magic = magic_operator.fit_transform(X, genes=['VIM', 'CDH1', 'ZEB1']) >>> X_magic.shape (500, 3) >>> magic_operator.set_params(t=7) MAGIC(a=15, k=5, knn_dist='euclidean', n_jobs=1, n_pca=100, random_state=None, t=7, verbose=1) >>> X_magic = magic_operator.transform(genes=['VIM', 'CDH1', 'ZEB1']) >>> X_magic.shape (500, 3) >>> X_magic = magic_operator.transform(genes="all_genes") >>> X_magic.shape (500, 197) >>> plt.scatter(X_magic['VIM'], X_magic['CDH1'], ... c=X_magic['ZEB1'], s=1, cmap='inferno') >>> plt.show() >>> magic.plot.animate_magic(X, gene_x='VIM', gene_y='CDH1', ... gene_color='ZEB1', operator=magic_operator) >>> dremi = magic_operator.knnDREMI('VIM', 'CDH1', plot=True)
References
[1] (1, 2, 3) Van Dijk D et al. (2018), Recovering Gene Interactions from Single-Cell Data Using Data Diffusion, Cell. -
diff_op The diffusion operator calculated from the data
-
fit(X, graph=None)[source] Computes the diffusion operator
Parameters: - X (array, shape=[n_samples, n_features]) – input data with n_samples samples and n_features dimensions. Accepted data types: numpy.ndarray, scipy.sparse.spmatrix, pd.DataFrame, anndata.AnnData.
- graph (graphtools.Graph, optional (default: None)) – If given, provides a precomputed kernel matrix with which to perform diffusion.
Returns: magic_operator – The estimator object
Return type:
-
fit_transform(X, graph=None, **kwargs)[source] Computes the diffusion operator and the denoised gene expression
Parameters: - X (array, shape=[n_samples, n_features]) – input data with n_samples samples and n_features dimensions. Accepted data types: numpy.ndarray, scipy.sparse.spmatrix, pd.DataFrame, anndata.AnnData.
- graph (graphtools.Graph, optional (default: None)) – If given, provides a precomputed kernel matrix with which to perform diffusion.
- genes (list or {"all_genes", "pca_only"}, optional (default: None)) – List of genes, either as integer indices or column names if input data is a pandas DataFrame. If “all_genes”, the entire smoothed matrix is returned. If “pca_only”, PCA on the smoothed data is returned. If None, the entire matrix is also returned, but a warning may be raised if the resultant matrix is very large.
- t_max (int, optional, default: 20) – maximum t to test if t is set to ‘auto’
- plot_optimal_t (boolean, optional, default: False) – If true and t is set to ‘auto’, plot the disparity used to select t
- ax (matplotlib.axes.Axes, optional) – If given and plot_optimal_t is true, plot will be drawn on the given axis.
Returns: X_magic – The gene expression values after diffusion
Return type: array, shape=[n_samples, n_genes]
-
knnDREMI(gene_x, gene_y, k=10, n_bins=20, n_mesh=3, n_jobs=1, plot=False, **kwargs)[source] Calculate kNN-DREMI on MAGIC output
Calculates k-Nearest Neighbor conditional Density Resampled Estimate of Mutual Information as defined in Van Dijk et al, 2018. [1]
Note that kNN-DREMI, like Mutual Information and DREMI, is not symmetric. Here we are estimating I(Y|X).
Parameters: - gene_x (array-like, shape=[n_samples]) – Gene shown on the x axis (independent feature)
- gene_y (array-like, shape=[n_samples]) – Gene shown on the y axis (dependent feature)
- k (int, range=[0:n_samples), optional (default: 10)) – Number of neighbors
- n_bins (int, range=[0:inf), optional (default: 20)) – Number of bins for density resampling
- n_mesh (int, range=[0:inf), optional (default: 3)) – In each bin, density will be calculcated around (mesh ** 2) points
- n_jobs (int, optional (default: 1)) – Number of threads used for kNN calculation
- plot (bool, optional (default: False)) – If True, DREMI create plots of the data like those seen in Fig 5C/D of van Dijk et al. 2018. (doi:10.1016/j.cell.2018.05.061).
- **kwargs (additional arguments for scprep.stats.plot_knnDREMI) –
Returns: dremi – kNN condtional Density resampled estimate of mutual information
Return type: float
-
set_params(**params)[source] Set the parameters on this estimator.
Any parameters not given as named arguments will be left at their current value.
Parameters: - knn (int, optional, default: 5) – number of nearest neighbors on which to build kernel
- decay (int, optional, default: 1) – sets decay rate of kernel tails. If None, alpha decaying kernel is not used
- t (int, optional, default: 3) – power to which the diffusion operator is powered. This sets the level of diffusion. If ‘auto’, t is selected according to the R squared of the diffused data
- n_pca (int, optional, default: 100) – Number of principal components to use for calculating neighborhoods. For extremely large datasets, using n_pca < 20 allows neighborhoods to be calculated in roughly log(n_samples) time.
- knn_dist (string, optional, default: 'euclidean') – recommended values: ‘euclidean’, ‘cosine’ Any metric from scipy.spatial.distance can be used distance metric for building kNN graph.
- n_jobs (integer, optional, default: 1) – The number of jobs to use for the computation. If -1 all CPUs are used. If 1 is given, no parallel computing code is used at all, which is useful for debugging. For n_jobs below -1, (n_cpus + 1 + n_jobs) are used. Thus for n_jobs = -2, all CPUs but one are used
- random_state (integer or numpy.RandomState, optional, default: None) – The generator used to initialize random PCA If an integer is given, it fixes the seed Defaults to the global numpy random number generator
- verbose (int or boolean, optional (default: 1)) – If True or > 0, print status messages
Returns: Return type: self
-
transform(X=None, genes=None, t_max=20, plot_optimal_t=False, ax=None)[source] Computes the values of genes after diffusion
Parameters: - X (array, optional, shape=[n_samples, n_features]) – input data with n_samples samples and n_features dimensions. Not required, since MAGIC does not embed cells not given in the input matrix to MAGIC.fit(). Accepted data types: numpy.ndarray, scipy.sparse.spmatrix, pd.DataFrame, anndata.AnnData.
- genes (list or {"all_genes", "pca_only"}, optional (default: None)) – List of genes, either as integer indices or column names if input data is a pandas DataFrame. If “all_genes”, the entire smoothed matrix is returned. If “pca_only”, PCA on the smoothed data is returned. If None, the entire matrix is also returned, but a warning may be raised if the resultant matrix is very large.
- t_max (int, optional, default: 20) – maximum t to test if t is set to ‘auto’
- plot_optimal_t (boolean, optional, default: False) – If true and t is set to ‘auto’, plot the disparity used to select t
- ax (matplotlib.axes.Axes, optional) – If given and plot_optimal_t is true, plot will be drawn on the given axis.
Returns: X_magic – The gene expression values after diffusion
Return type: array, shape=[n_samples, n_genes]